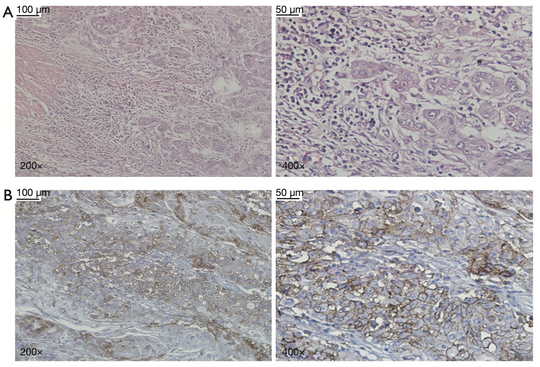
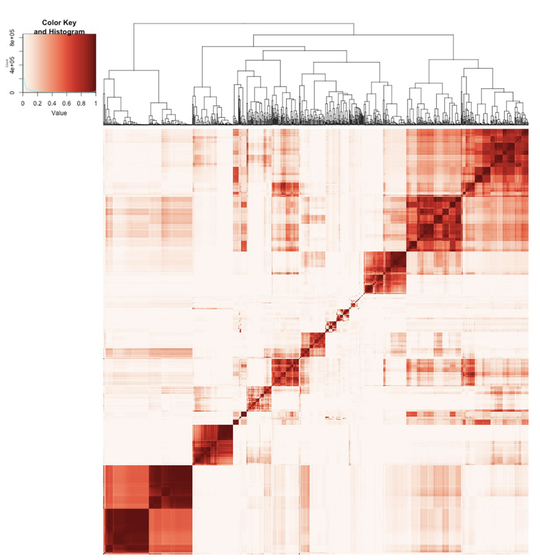
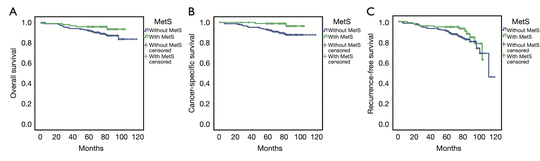
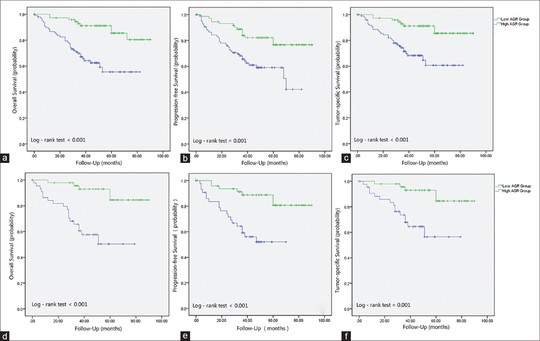
Sebastian (Shaobo) Li
PhD in Cancer Biology and Genomics
University of Southern California
About me
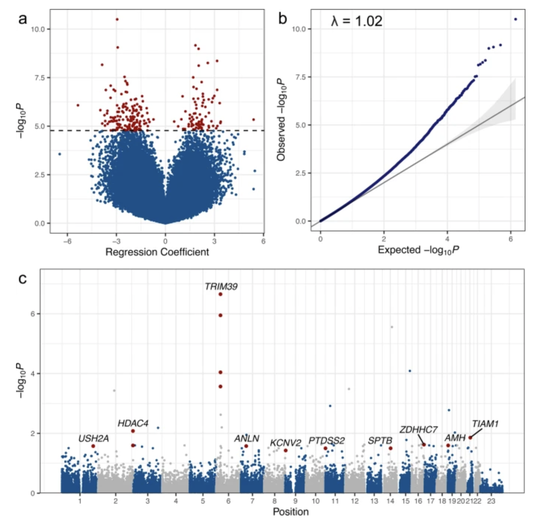
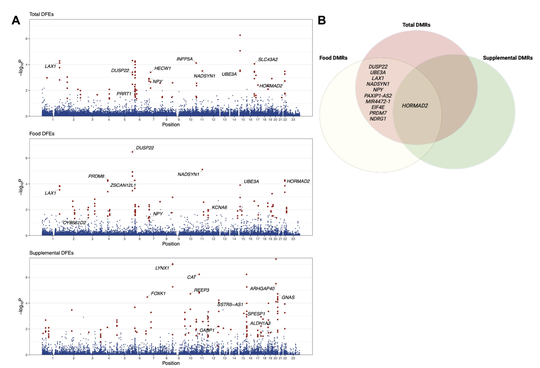
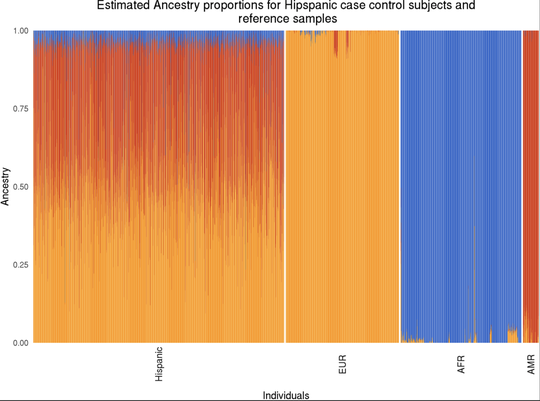
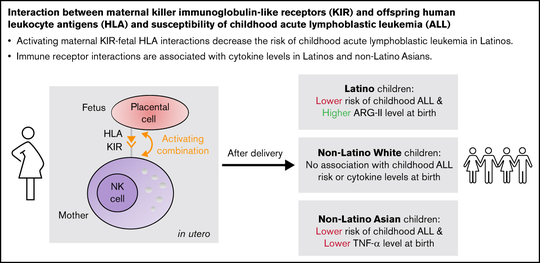
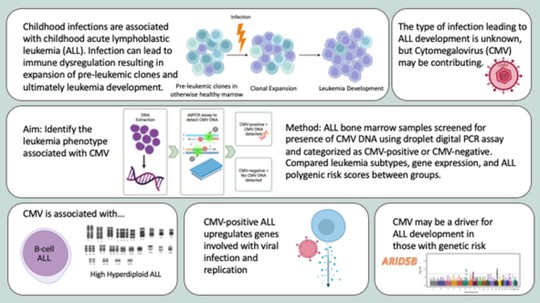
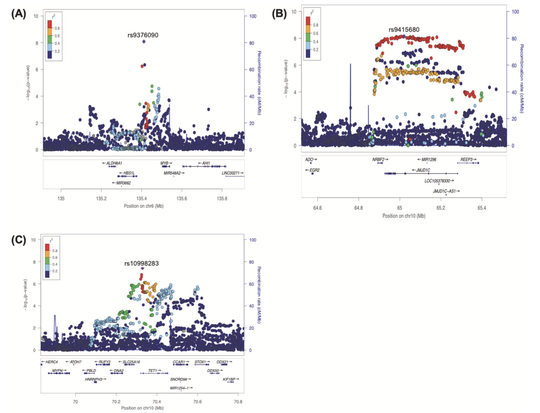
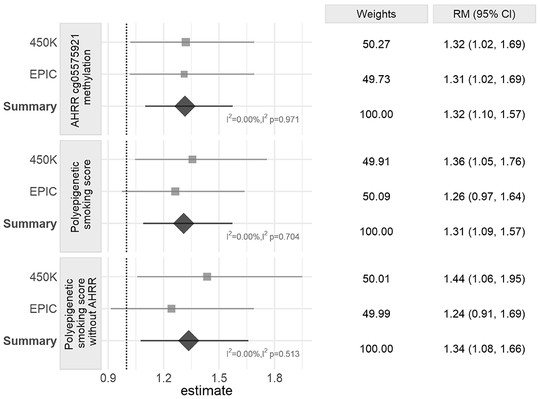
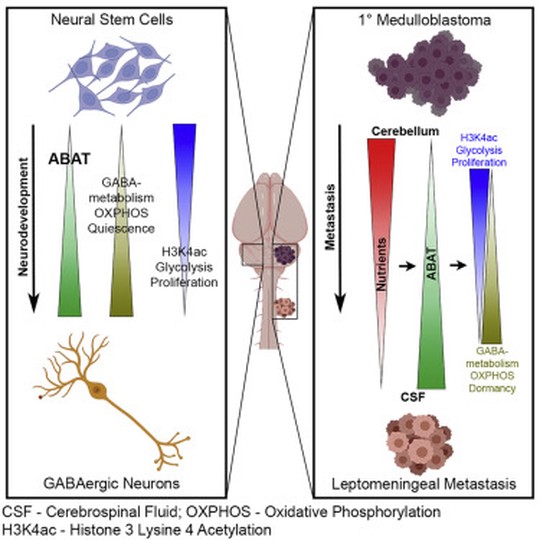
My name is Sebastian (Shaobo) Li. I graduated from the Cancer Biology and Genomics PhD program at the USC Keck School of Medicine.I was trained as a bioinformatician with a focus on genetic epidemiology. My research involves using genetic and epigenetic association analyses, ancestry analyses and machine learning/deep learning models to understand risks of childhood cancers.
I have a wide range of interests. Learning new tools, ideas and skills always excites me.
Interests
- Bioinformatics (genetic, epigenetic, ancestry analysis)
- Machine learning, deep learning
- Data science
Education
-
PhD in Cancer Biology and Genomics (Bioinformatics), 2018 – 2022
University of Southern California
-
Bachelor of Medicine, 2009 – 2014
Fudan University